
Types of RNP Approach
Before GPS became available there were two categories of instrument approach; Precision and Non-Precision. The advent of DGPS and PBN performance standards has introduced a new category of instrument approach; an Approach with Vertical Guidance (APV). The diagram below illustrates the types of approach that may be carried out using GNSS equipment and that are regulated by the PBN navigation specification for GNSS approaches (RNP APCH).
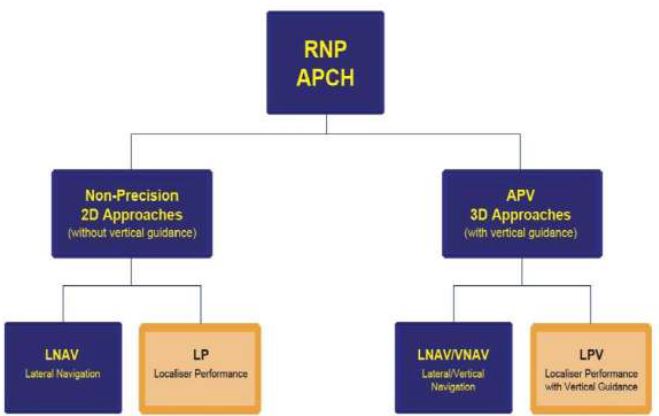
Approaches Without Vertical Guidance
Non-Precision Two Dimensional (2D) Approaches. There are two types of RNP approach that do not offer any vertical guidance information and therefore fall into the Non-Precision approach category. Approach minima for both types of these RNAV approaches are therefore either Minimum Descent Altitudes (MDA) or Minimum Descent Heights (MDH):
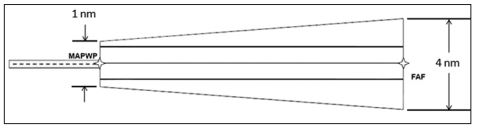
- Lateral Navigation (LNAV). This approach provides only lateral guidance in which the displayed centreline sensitivity remains linear as the aircraft approaches the runway. SBAS may be enabled for LNAV approaches, but is not mandatory; however, RAIM is mandatory if SBAS is unavailable. The bold lines on the diagram below indicate the aircraft position if the CDI indication remains just at maximum deflection throughout the approach:
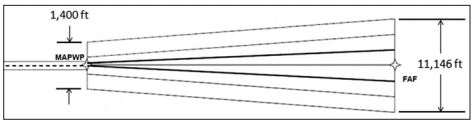
- Localiser Performance (LP). This approach provides only lateral guidance that is angular (like an ILS localiser) on the approach. The displayed centreline sensitivity increases as the aircraft approaches the runway. SBAS is mandatory. The bold lines on the diagram below indicate the aircraft position if the CDI indication remains just at the maximum deflection throughout the approach:
G1000 Lateral Navigation with Advisory Vertical Guidance Mode (LNAV+V) When SBAS is enabled, G1000 provides an advisory vertical guidance in association with LP or LNAV approaches. The intent is to aid the pilot in flying a CDFA path to the MDA/H. When this additional information is presented, the Flight Phase annunciation displays LNAV+V and the system displays artificially created advisory glide path information from the final approach fix to the touchdown point on the runway. LNAV+V is not a 3D approach, so if LNAV+V mode activates, you will still be flying a 2D, LP or LNAV, approach. Pilots must therefore use the barometric altimeter as the primary altitude reference to meet all altitude/range restrictions and treat the approach as either an LNAV or LP approach using MDA or MDH minima as appropriate. Advisory vertical guidance is not required and is an optional capability available only if SBAS is active.
Approaches With Vertical Guidance
Three Dimensional (3D) Approaches. There are two types of RNP approach that offer usable vertical guidance information. The approach minima used for both of these types of approach are either Decision Altitudes (DA) or Decision Heights (DH):
- Localiser Performance with Vertical Guidance (LPV). This approach provides both lateral and vertical guidance that is angular in nature (like an ILS). The displayed sensitivity of the centreline and glidepath increases as the aircraft approaches the runway. SBAS is mandatory as it ensures the GPS accuracy required to provide glidepath indications without use of any altimeter input. Like on a ILS approach, half-scale deflection (one dot) of the centreline and glidepath is the maximum permitted and a deviation beyond those limits requires the pilot to go-around.
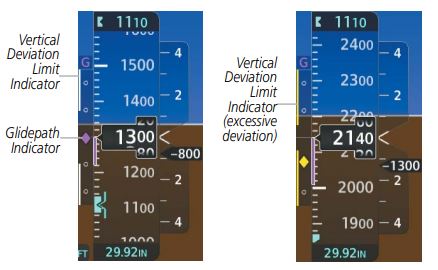
- Lateral Navigation and Vertical Navigation (LNAV/VNAV). This approach provides both lateral and vertical guidance, but similar to the LNAV approach, the displayed centreline sensitivity remains linear as the aircraft approaches the runway. Whilst the displayed Glidepath Indicator reacts in an angular fashion, like on an ILS glideslope, by definition the required vertical accuracy is a linear +/- 75ft throughout the approach, not a half-scale deflection. To allow the pilot to recognise an excessive vertical deviation, a pair Vertical Deviation Limit Indicators appear to the left of the glideslope when the aircraft passes the Final Approach Fix. The pilot must keep the Glidepath Indicator between then throughout the approach. Since the limits are linear, the gap between the Vertical Deviation Limit Indicators widens as the aircraft approaches the runway.
 The LNAV/VNAV approach may take one of these two forms dependant on the availability of SBAS:
The LNAV/VNAV approach may take one of these two forms dependant on the availability of SBAS:
-
- Barometric Vertical Guidance (Baro VNAV). If SBAS is not available, but RAIM is available (it is mandatory), glidepath information may be provided from a suitable aircraft altimetry system. Developed before SBAS and LPV approaches were introduced, this is a legacy approach mode developed for use by aircraft equipped with multiple, independent altimeter systems. The Seneca is not equipped for this type of approach so the option will not be offered. Warning! Crews carrying out this type of approach must ensure that the correct QNH and altimeter temperature error corrections are applied because altimeter errors will impact the glidepath displayed. If altimeter temperature error corrections cannot be applied in low temperatures, the approach may be prohibited.
- Lateral Navigation and Vertical Navigation using SBAS enhancement (L/VNAV). If SBAS is available, the GPS will provide glidepath information. RAIM is not required. Linear guidance is displayed for the centerline information, but although the glidepath indicator acts in an angular fashion, the Vertical Deviation Limit Indicators will display on passing the Final Approach Fix, as described above.
Click the video above to view a typical L/VNAV approach to RW23 at Cambridge filmed in the simulator. Notice how the Vertical Deviation Limit Indicators appear at the FAF and the gap between them widens as the approach progresses towards the MAP. […and also notice how the aircraft climbed 70ft when the student lowered the flaps just before the FAF. The student did not select a lower pitch attitude as the flaps travelled so the extra lift caused the aircraft to climb. Allowing that to happen (especially on a hot day) can put the aircraft above the vertical deviation limit as soon as the Final Approach starts! — Ed]
-
Instrument Approach Minima
This is the latest extract from EASA regulations (NCO.OP.111) which outlines the lowest possible DH/MDH for instrument procedures:
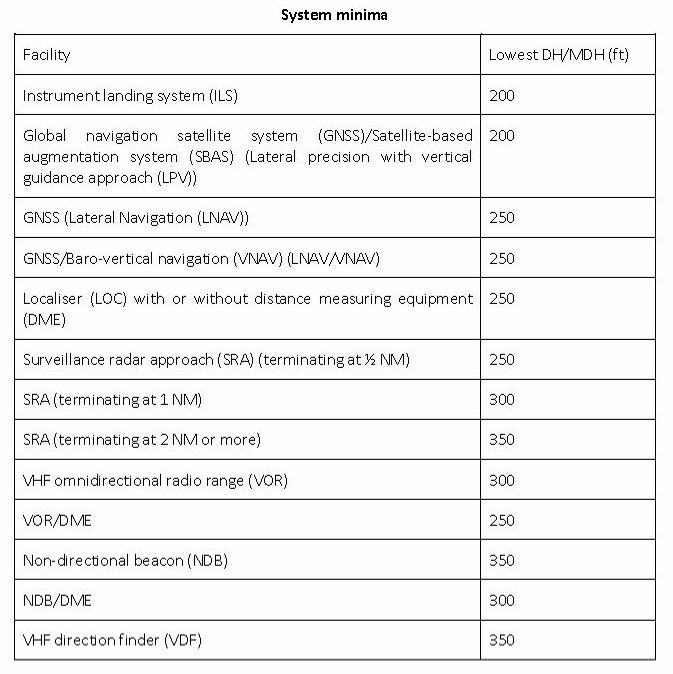
It can be seen that the lowest permissible MDH for GNSS approaches without approved vertical guidance is 250ft. Originally, LPV approaches also had a limit of 250ft, but introduction of the new LPV-200 specification now allows a limit of 200ft to be used for those approaches surveyed to those standards.














